Polygenic risk scores (PRS) estimate an individual's genetic likelihood of complex traits and diseases by aggregating information across multiple genetic variants identified from genome-wide association studi...
详细信息
Polygenic risk scores (PRS) estimate an individual's genetic likelihood of complex traits and diseases by aggregating information across multiple genetic variants identified from genome-wide association studies. PRS can predict a broad spectrum of diseases and have therefore been widely used in research settings. Some work has investigated their potential applications as biomarkers in preventative medicine, but significant work is still needed to definitively establish and communicate absolute risk to patients for genetic and modifiable risk factors across demographic groups. However, the biggest limitation of PRS currently is that they show poor generalizability across diverse ancestries and cohorts. Major efforts are underway through methodological development and data generation initiatives to improve their generalizability. This review aims to comprehensively discuss current progress on the development of PRS, the factors that affect their generalizability, and promising areas for improving their accuracy, portability, and implementation.
The All of Us Research program's Data and Research Center (DRC) was established to help acquire, curate, and provide access to one of the world's largest and most diverse datasets for precision medic...
详细信息
The All of Us Research program's Data and Research Center (DRC) was established to help acquire, curate, and provide access to one of the world's largest and most diverse datasets for precision medicine research. Already, over 500,000 participants are enrolled in All of Us, 80% of whom are underrepresented in biomedical research, and data are being analyzed by a community of over 2,300 researchers. The DRC created this thriving data ecosystem by collaborating with engaged participants, innovative program partners, and empowered researchers. In this review, we first describe how the DRC is organized to meet the needs of this broad group of stakeholders. We then outline guiding principles, common challenges, and innovative approaches used to build the All of Us data ecosystem. Finally, we share lessons learned to help others navigate important decisions and trade-offs in building a modern biomedical data platform.
Advances in single-cell proteomics technologies have resulted in high-dimensional datasets comprising millions of cells that are capable of answering key questions about biology and disease. The advent of these techno...
Advances in single-cell proteomics technologies have resulted in high-dimensional datasets comprising millions of cells that are capable of answering key questions about biology and disease. The advent of these technologies has prompted the development of computational tools to process and visualize the complex data. In this review, we outline the steps of single-cell and spatial proteomics analysis pipelines. In addition to describing available methods, we highlight benchmarking studies that have identified advantages and pitfalls of the currently available computational toolkits. As these technologies continue to advance, robust analysis tools should be developed in tandem to take full advantage of the potential biological insights provided by these data.
Recent advancements in single-cell technologies have enabled expression quantitative trait locus (eQTL) analysis across many individuals at single-cell resolution. Compared with bulk RNA sequencing, which averages gen...
Recent advancements in single-cell technologies have enabled expression quantitative trait locus (eQTL) analysis across many individuals at single-cell resolution. Compared with bulk RNA sequencing, which averages gene expression across cell types and cell states, single-cell assays capture the transcriptional states of individual cells, including fine-grained, transient, and difficult-to-isolate populations at unprecedented scale and resolution. Single-cell eQTL (sc-eQTL) mapping can identify context-dependent eQTLs that vary with cell states, including some that colocalize with disease variants identified in genome-wide association studies. By uncovering the precise contexts in which these eQTLs act, single-cell approaches can unveil previously hidden regulatory effects and pinpoint important cell states underlying molecular mechanisms of disease. Here, we present an overview of recently deployed experimental designs in sc-eQTL studies. In the process, we consider the influence of study design choices such as cohort, cell states, and ex vivo perturbations. We then discuss current methodologies, modeling approaches, and technical challenges as well as future opportunities and applications.
In clinical artificial intelligence (AI), graph representation learning, mainly through graph neural networks and graph transformer architectures, stands out for its capability to capture intricate relationships and s...
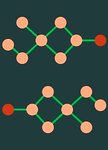
In clinical artificial intelligence (AI), graph representation learning, mainly through graph neural networks and graph transformer architectures, stands out for its capability to capture intricate relationships and structures within clinical datasets. With diverse data—from patient records to imaging—graph AI models process data holistically by viewing modalities and entities within them as nodes interconnected by their relationships. Graph AI facilitates model transfer across clinical tasks, enabling models to generalize across patient populations without additional parameters and with minimal to no retraining. However, the importance of human-centered design and model interpretability in clinical decision-making cannot be overstated. Since graph AI models capture information through localized neural transformations defined on relational datasets, they offer both an opportunity and a challenge in elucidating model rationale. Knowledge graphs can enhance interpretability by aligning model-driven insights with medical knowledge. Emerging graph AI models integrate diverse data modalities through pretraining, facilitate interactive feedback loops, and foster human–AI collaboration, paving the way toward clinically meaningful predictions.
Analysis of cancer genomes has shown that a large fraction of chromosomal changes originate from catastrophic events including whole-genome duplication, chromothripsis, breakage-fusion-bridge cycles, and chromoplexy. ...
详细信息
Analysis of cancer genomes has shown that a large fraction of chromosomal changes originate from catastrophic events including whole-genome duplication, chromothripsis, breakage-fusion-bridge cycles, and chromoplexy. Through sophisticated computational analysis of cancer genomes and experimental recapitulation of these catastrophic alterations, we have gained significant insights into the origin, mechanism, and evolutionary dynamics of cancer genome complexity. In this review, we summarize this progress and survey the major unresolved questions, with particular emphasis on the relative contributions of chromosome fragmentation and DNA replication errors to complex chromosomal alterations.







暂无评论